Study shows that Rett syndrome in females is not just less severe, but different
By Nadine A Yehya
A long-term study on mice explains why the condition progresses differently in males and females
(SACRAMENTO)
A new UC Davis MIND Institute study offers critical insights into Rett syndrome, a rare genetic condition that affects mostly girls. The research reveals how this condition affects males and females differently, with symptom progression linked to changes in gene responses in brain cells.
Rett syndrome is caused by mutations of the MECP2 gene located on the X chromosome. Children with Rett initially show typical development before symptoms start.
The symptoms vary widely. They include loss of hand function, breathing difficulties and seizures that affect the child’s ability to speak, walk and eat. Rett is less common in males, but when they are affected, symptoms are typically more severe and present earlier than in females.
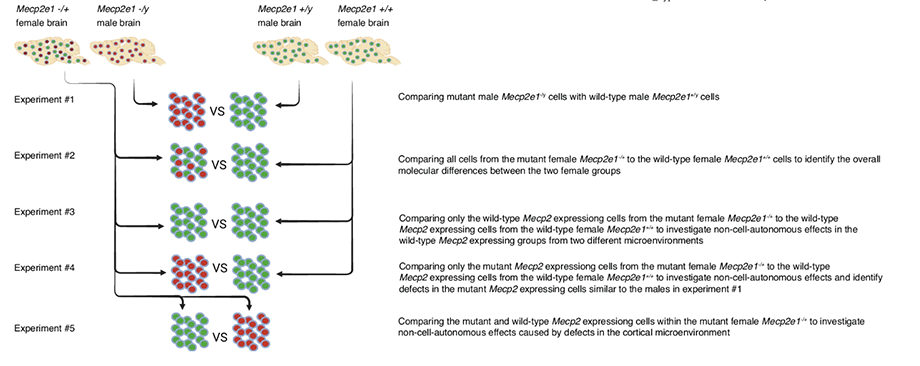
The study, published in Communications Biology, analyzed the cerebral cortices of male and female mice with and without MECP2 mutation at three timepoints: before symptoms, when symptoms started, and during late disease stage. It looked at gene expression in 14 different cell types.
“In an X-linked dominant disorder such as Rett, it is important to know that females do not just have less severe symptoms than males. Their condition is different,” said Janine LaSalle, lead author of the study and professor of microbiology and immunology at UC Davis Health. “That is why it is important to study female mouse models of Rett. They are more relevant for developing treatments.”
It is important to study female mouse models of Rett. They are more relevant for developing treatments.” —Janine LaSalle, professor of microbiology and immunology
Most studies use male mouse models for Rett syndrome. These models have an engineered deletion of key MECP2 gene elements. This disables the production of MeCP2 protein in all cells, as males have only one X chromosome.
In humans with Rett, this type of MECP2 gene deletion does not exist. All the cells in females with Rett have an MECP2 mutation inherited from one parent on the X chromosome, but only half of the cells express the mutant gene. This means the other half of the cells with a wild-type copy of MECP2 inherited from the other parent expresses normal MeCP2 protein.
Thus, the brain of a girl with Rett syndrome has a mosaic-like distribution of cells expressing wild-type MeCP2 and cells expressing mutant MeCP2 protein.
“When we separated the two types of cells in the brain, we could see that the wild-type expressing cells are really dysregulated,” LaSalle said.
The study design with five experiments.
Mosaic brains and the seesaw effect in dysregulated genes
Gene regulation is the process of turning genes on and off. Dysregulated genes are either over expressing or under expressing, which means they are producing more or less of certain functional proteins. In the female mouse model with Rett syndrome, this gene impairment went in stages. The researchers called this “the seesaw effect.”
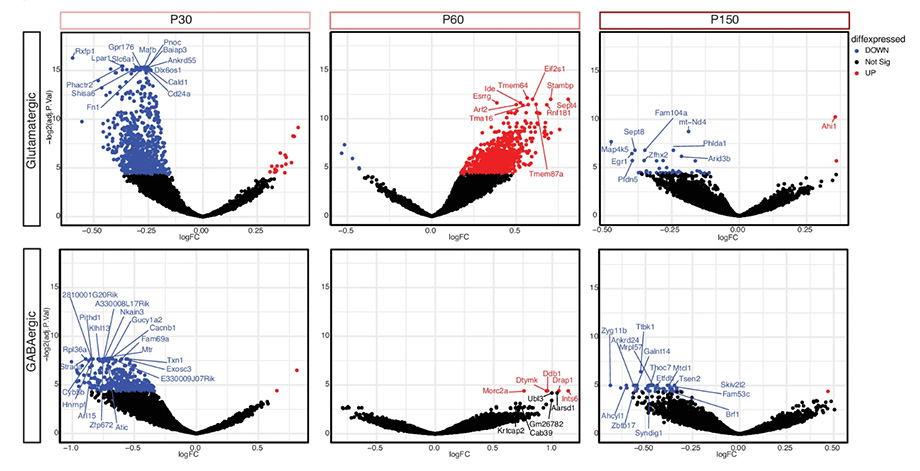
The authors studied the wild-type expressing cells of mosaic female Rett mouse brain. They found changes to gene responses in excitatory neurons before symptoms started, in inhibitory neurons when symptoms started, and then in astrocytes at a later stage.
“There was a back-and-forth oscillation where it seems like the genes are trying to reach homeostasis or balance in the brain. This idea of the seesaw homeostasis is important to look at over time, as the symptoms progress,” LaSalle said.
LaSalle explained that the wild-type expressing cells are trying to counteract the effects from the mutant expressing cells. By doing so, they become dysregulated themselves. The wild-type cells start expressing genes up and down. This dysregulation is worse in the beginning and then it stabilizes as Rett progresses.
An example of the seesaw in gene response changes over time. Blue are genes that are down, red are genes that are up in either excitatory (top) or inhibitory (bottom) responses over three time points: P30 (pre-symptomatic), P60 (early symptomatic), or P150 (late symptomatic) in female brain from Rett syndrome model compared to controls.
Finding balance as disease progresses
The study found an unusual disease progression. One can start out with normal infancy, regress, then plateau, and then regress again and then finally plateau. Plateauing resembles a state of homeostasis, a stabilization of symptoms over time.
“We expected the number of impaired genes to go along with the increased symptoms. To our surprise, the females actually had more dysregulated genes at the pre-symptomatic stage than they did later on,” LaSalle said.
The study also found that females overall had more dysregulated genes and at different disease stages compared to males. This suggests that males are not just experiencing a more severe version of Rett. Importantly, this study confirmed that the use of a female mouse model is a better representation of Rett syndrome in females.
The study also examined various gene pathways. A pathway is like taking a diverse group of genes and putting them into a functional group. If the human body is a factory, a pathway would be a team within the factory. For example, a team would be keeping the heart rate normal. Another would be working on the sleep-wake cycle.
The study showed a link between the MECP2 mutation and the Alzheimer’s pathway as well as various addiction pathways.
“In genomics, we have moved from looking at one gene at a time to thinking about groups of genes that act together in pathways. The findings point to different pathways, which means MECP2 mutation may have relevance to other diseases beyond Rett syndrome,” LaSalle explained.
Members of the LaSalle Lab
This study was supported by multiple National Institutes of Health grants (1R01AA027075, 1S10OD010786-01, P50 HD103526, P30 ES023513), the National Cancer Institute grant (P30 CA093373) and Astrios Cell Sorter grant (S10 OD018223). UC Davis co-authors include Osman Sharifi, Dag Yasui, Viktoria Haghani, Kari Neier, Sophia Hakam, Keith Fraga, Ian Korf, Gerald Quon and Nelson Johansen.
If you have serious symptoms of illness, contact your primary care provider. UC Davis Health patients can use the MyUCDavisHealth symptom tracker to evaluate whether to seek help. Telehealth video visits and Express Care are also available.
If you have a medical emergency, call 911 and notify them of your COVID-19 symptoms.
Ways to Seek Treatment
If you test positive for COVID-19 at home, you can contact your primary care provider about a prescription for Paxlovid.