


Raymond Gong, MD; Karen Matsukuma, MD PhD
Introduction
Colorectal cancer is the third most common cause of cancer death in both men and women in the United States [1]. The vast majority of colorectal cancers are adenocarcinomas, which are malignancies arising from the epithelial cells of colorectal mucosa. The transformation from normal colonic epithelium to adenocarcinoma is driven by certain genetic mutations which can be acquired or inherited. The study of these mutations has led to the identification of unique molecular pathways and has uncovered implications for cancer prognosis and treatment.
Overview of Molecular Pathways and their Clinical Significance
Established molecular pathways in colorectal tumorigenesis include chromosomal instability, DNA mismatch repair deficiency, and DNA hypermethylation. Chromosomal instability results from the stepwise accumulation of mutations, as seen in the adenoma-carcinoma sequence typical of most sporadic colorectal adenocarcinomas and in familial adenomatous polyposis [2]. Mutations result in activation of growth promoting pathways (e.g., APC gene mutations, which occur early in the process) and diminished activity of apoptotic pathways and tumor suppressor genes (e.g., KRAS mutations and TP53 mutations).
DNA mismatch repair deficiency is the basis of Lynch syndrome (previously known as hereditary nonpolyposis colorectal cancer) as well as approximately 15 percent of sporadic colorectal adenocarcinomas [3]. This pathway is characterized by mutations in the DNA mismatch repair (MMR) enzymes genes, MLH1, PMS2, MSH2, and MSH6. These mutations lead to impaired DNA repair capacity and the accumulation of DNA replication errors. Microsatellites, short repetitive non-coding sequences of DNA scattered throughout the genome, are particularly vulnerable to such errors, which result in an altered length of the DNA microsatellite sequences (a phenomenon known as microsatellite instability, MSI). Tumors with defects in the DNA MMR pathway are characterized by high levels of microsatellite instability (MSI-H).
Colorectal adenocarcinomas with DNA hypermethylation are characterized by a high frequency of epigenetic DNA methylation of CpG islands and are referred to as having the CpG island methylator phenotype, or CIMP+ [4]. In many cases, hypermethylation of the MLH1 gene leads to gene silencing and subsequent DNA MMR deficiency. BRAF mutations (most commonly in the V600E codon) are strongly associated with CIMP+, MSI-H sporadic colorectal adenocarcinomas, are mutually exclusive with KRAS mutations, and are very rarely seen in Lynch syndrome-related tumors [5].
The above pathways include molecular markers that are prognostic and predictive. In particular, MSI-H colorectal adenocarcinoma in patients with localized disease has been associated with longer survival than DNA MMR stable tumors [6]. Treatment options may also be affected, as patients with MSI-H tumors are less likely to benefit from fluorouracil-based adjuvant chemotherapy and instead may benefit from checkpoint inhibitor immunotherapy (in the setting of advanced disease) [6-8]. In comparison to MSI-H tumors, microsatellite stable tumors with BRAF and KRAS mutations are consistently associated with worse prognosis, and these mutations may confer less responsiveness to anti-EGFR therapy in advanced disease [9-11].
Laboratory Best Practice for Routine Mismatch Repair Deficiency Testing
At UC Davis Health, all colorectal biopsy and resection specimens with a new diagnosis of primary adenocarcinoma undergo DNA MMR deficiency testing, in accordance with established guidelines on molecular markers for the evaluation of colorectal cancer [12]. Universal testing for DNA MMR deficiency is performed for identification of patients at increased risk for Lynch syndrome but also confers prognostic information and may be therapeutically relevant.
The following flowchart summarizes the algorithm employed at UC Davis Health.
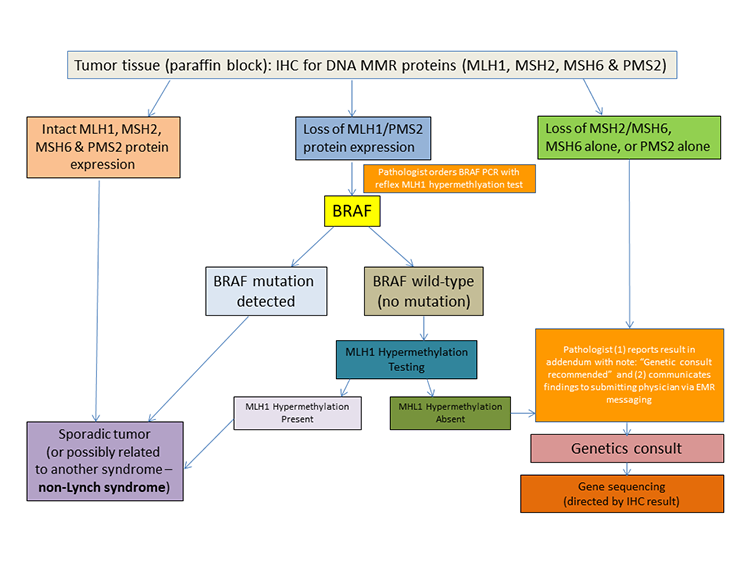
DNA MMR deficiency is evaluated by immunohistochemistry (IHC) using a panel of stains for the DNA MMR proteins MLH1, MSH2, MSH6, and PMS2 on formalin-fixed paraffin-embedded tissue containing colorectal adenocarcinoma. IHC is an indirect but reliable and cost-effective first-line screen for DNA MMR deficiency [13]. Of note, this panel will miss rare cases caused by mutations in other genes involved in DNA MMR deficiency (e.g., EPCAM).
The IHC slides are reviewed by a gastrointestinal pathologist to evaluate for nuclear expression in the tumor cells. Absence of nuclear expression in tumor cells, with retained nuclear expression in the background stromal cells (internal control), is considered an abnormal result. In the DNA MMR pathway, MLH1 forms a heterodimer with PMS2, and MSH2 forms a heterodimer with MSH6. Loss of MLH1 leads to protein degradation of PMS2 (the dependent partner), and thus MLH1 and PMS2 nuclear expression is typically lost together. However, if a gene defect in PMS2 results in loss of PMS2 expression, MLH1 protein expression is typically retained. Similarly, loss of MSH2 (dominant partner) leads to loss of MSH6, whereas loss of MSH6 does not lead to degradation of MSH2.
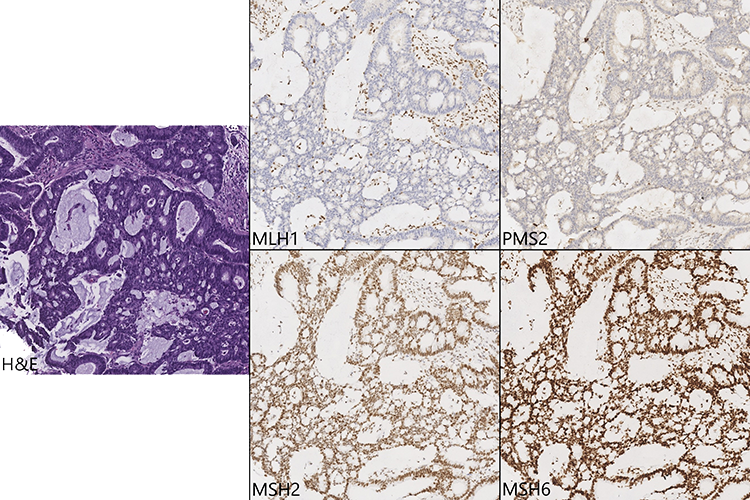
The following figure shows a colonic adenocarcinoma (image of hematoxylin and eosin (H&E)-stained slide provided for reference) demonstrating a typical pattern of MLH1, and, consequently, PMS2 loss with intact expression of MSH2 and MSH6. In addition to external positive controls, stromal cells and/or other benign components (e.g., vessels, lymphocytes) present alongside the invasive adenocarcinoma serve as internal positive controls.
As seen in the rightmost arm of the algorithm flowchart above, the loss of MSH2 and MSH6 together, MSH6 alone, or PMS2 alone are results which are strongly suggestive of Lynch syndrome. Such results are immediately communicated to the clinician so that a genetics consultation can be offered.
When loss of MLH1 expression is detected, as seen in the middle arm of the algorithm flowchart, sequencing of BRAF codon 600 by PCR is reflexively ordered by the pathologist. If a mutation in BRAF codon 600 is detected, the result essentially excludes the possibility of a Lynch syndrome-associated colorectal carcinoma, and no further testing is performed. However, if no BRAF codon 600 mutation is found, reflex testing for MLH1 promoter hypermethylation is performed. If no evidence of MLH1 promoter hypermethylation is found, the likelihood of a germline MLH1 mutation (Lynch syndrome) is increased, and genetic counseling is recommended.
Conclusion
The study of molecular pathogenesis in colorectal adenocarcinoma has resulted in the identification of pathways and markers that have been incorporated into clinical care guidelines. Universal DNA MMR screening for all colorectal adenocarcinomas is currently the standard of care at most large institutions, including UC Davis Health, and is supported by multiple national organizations (National Comprehensive Cancer Network, U.S. Multi-Society Task Force on Colorectal Cancer, American Society of Clinical Oncology, American College of Gastroenterology).
References
- Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA Cancer J Clin. 2021;71(1):7-33.
- Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61(5):759-767.
- Shibata D, Peinado MA, Ionov Y, Malkhosyan S, Perucho M. Genomic instability in repeated sequences is an early somatic event in colorectal tumorigenesis that persists after transformation. Nat Genet. 1994;6(3):273-281.
- Weisenberger DJ, Siegmund KD, Campan M, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006;38(7):787-793.
- Domingo E, Niessen RC, Oliveira C, et al. BRAF-V600E is not involved in the colorectal tumorigenesis of HNPCC in patients with functional MLH1 and MSH2 genes. Oncogene. 2005;24(24):3995-3998.
- Popat S, Hubner R, Houlston RS. Systematic review of microsatellite instability and colorectal cancer prognosis. J Clin Oncol. 2005;23(3):609-618.
- Sargent DJ, Marsoni S, Monges G, et al. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J Clin Oncol. 2010;28(20):3219-3226.
- Le DT, Uram JN, Wang H, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med. 2015;372(26):2509-20.
- Modest DP, Ricard I, Heinemann V, et al. Outcome according to KRAS-, NRAS- and BRAF-mutation as well as KRAS mutation variants: pooled analysis of five randomized trials in metastatic colorectal cancer by the AIO colorectal cancer study group. Ann Oncol. 2016;27(9):1746-1753.
- Taieb J, Zaanan A, Le Malicot K, et al. Prognostic effect of BRAF and KRAS mutations in patients with stage III colon cancer treated with leucovorin, fluorouracil, and oxaliplatin with or without cetuximab: a post hoc analysis of the PETACC-8 trial. JAMA Oncol. 2016;2(5):643-653.
- Taieb J, Shi Q, Pederson L, et al. Prognosis of microsatellite instability and/or mismatch repair deficiency stage III colon cancer patients after disease recurrence following adjuvant treatment: results of an ACCENT pooled analysis of seven studies. Ann Oncol. 2019;30(9):1466-1471.
- Sepulveda AR, Hamilton SR, Allegra CJ, et al. Molecular biomarkers for the evaluation of colorectal cancer: guideline from the American Society for Clinical Pathology, College of American Pathologists, Association for Molecular Pathology, and the American Society of Clinical Oncology. J Clin Oncol. 2017;35(13):1453-1486.
- Fleming M, Ravula S, Tatishchev SF, Wang HL. Colorectal carcinoma: Pathologic aspects. J Gastrointest Oncol. 2012;3(3):153-173.
