
Case of the Month
October 2016 - Presented by Dr. Saba Ali & Dr. Denis Dwyre
Discussion
This patient presents with a history of abnormal bruising and bleeding suggestive of platelet abnormality. The abnormal laboratory findings including decreased von Willebrand factor activity: von Willebrand factor (vWF) antigen ratio (<1), and decreased ristocetin-induced platelet aggregation (RIPA), suggestive of a qualitative platelet abnormality. The next step in the work-up of this patient is a VWF multimeric analysis in order to distinguish the type of VWD. In this patient, multimeric analysis was normal. The vWF and multimer analysis findings are most consistent with von Willebrand disease, type 2M.
“Von Willebrand disease is an inherited bleeding disorder that is caused by deficiency or dysfunction of von Willebrand factor, a plasma protein that mediates the initial adhesion of platelets at sites of vascular injury and also binds and stabilized blood clotting factor VIII (FVIII) in the circulation.”1 The prevalence of vWD is approximately 0.8 percent.
VWF is a protein that circulates in the plasma as a protein structurally consisting of multiple identical subunits of varying size arranged in a linear pattern. VWF is synthesized in the vascular endothelial cells and megakaryocytes in the bone marrow. In the endothelium, VWF is stored in Wiebel-Palade bodies, secretory granules from which the protein is released in response to stress and/or drugs, such as desmopressin. Within the megakaryocytes, VWF is stored in platelet alpha-granules from which it is released following platelet aggregation. The major role of VWF is defined in primary hemostasis. Vascular injury causing exposure of the subendothelial collagen results in platelets binding to VWF via glycoprotein 1B/V/IX receptors. Lack of either VWF or the platelet receptor can result in deficiencies of the primary hemostatic plug and lead to bleeding clinically manifested in the mucosa, joints, or skin as bruising.
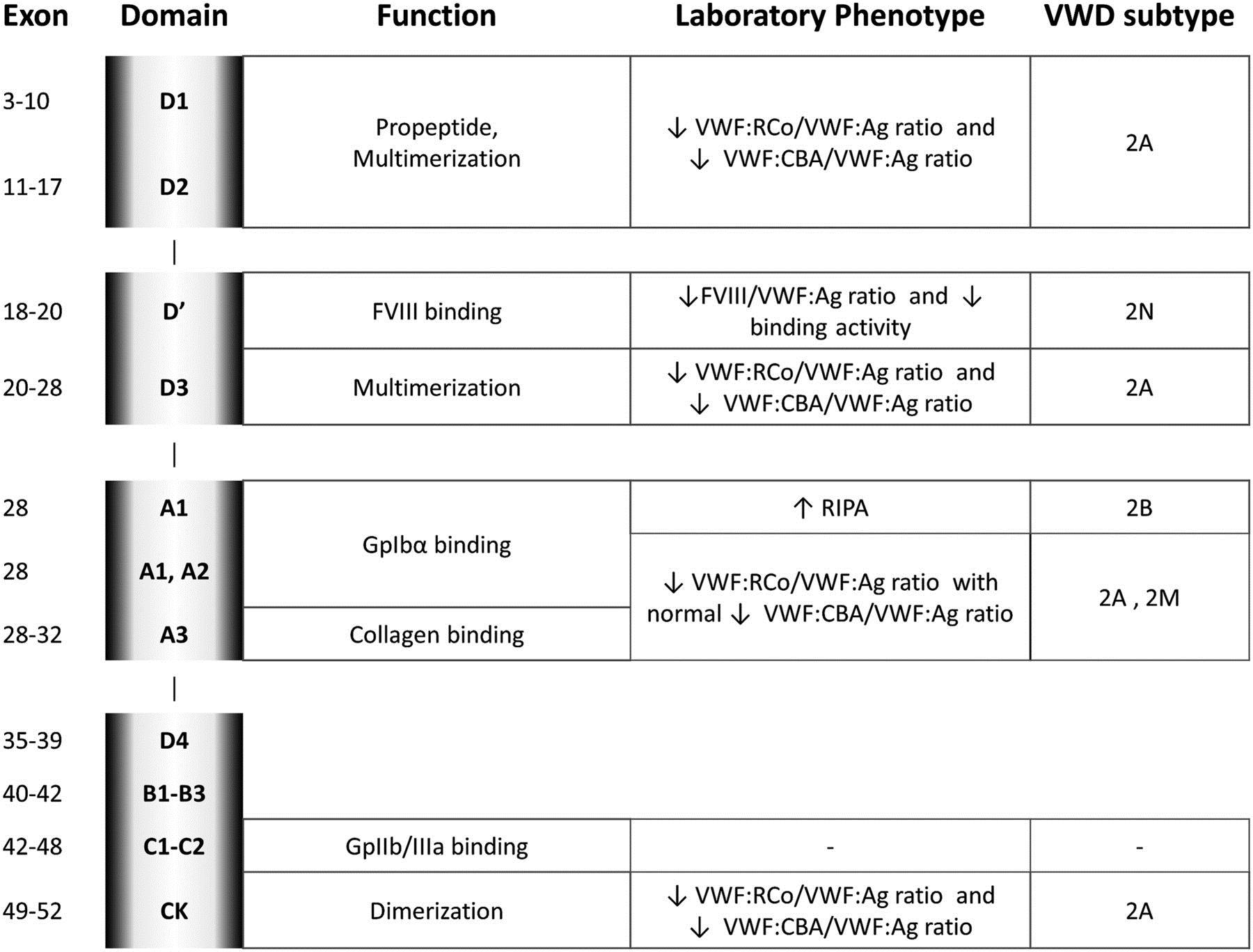
There are three main subtypes of VWD are depicted in table 1. VWF molecular abnormalities, protein domain localization, laboratory phenotype, and subtype classification in type 2 VWD is shown in table 2.
The patient described above has the characteristic clinical findings of type 2M VWD. This is an uncommon, mostly autosomal dominant disorder characterized by reduced binding of VWF to glycoprotein 1B platelet receptors, resulting from a loss-of-function mutation at the VWF binding site. Affected patients will typically have laboratory findings that show:
- A personal or family history of mucosal bleeding
- Ristocetin cofactor reduced vWF: Ag ratio (<0.5-0.7)
- Normal spectrum of VWF multimers
- RIPA impairment
Molecular studies in type 2M VWD occur in exon 28 with few recurrent muations known.
Antihemophilic Factor/von Willebrand Factor Complex (Humate-P) is used in the treatment of patients with hemophilia A and adults and pediatric patients with VWD during episodes of bleeding (due to trauma, surgery, or spontaneous events).
Table 1

Table 2
Alberto Tosetto and Giancarlo Castaman. How I treat type 2 variant forms of von Willebrand disease. Blood. 2015;125(6):907-914.
Resources
- Alberto Tosetto and Giancarlo Castaman. How I treat type 2 variant forms of von Willebrand disease. Blood. 2015;125(6):907-914)
- Sadler JE (1994). "A revised classification of von Willebrand disease. For the Subcommittee on von Willebrand Factor of the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis". Thromb. Haemost. 71 (4): 520 5.
- Clinical presentation and diagnosis of von Willebrand disease. http://www.uptodate.com/home/index.html
- Sadler JE, Budde U, Eikenboom JC, Favaloro EJ, Hill FG, Holmberg L, Ingerslev J, Lee CA, Lillicrap D, Mannucci PM, et al. Update on the pathophysiology and classification of von Willebrand disease: a report of the Subcommittee on von Willebrand Factor. J Thromb Haemost 2006 Oct;4(10):2103–2114.
- http://www.humate-p.com/Professional/What-is-VWD/Default.aspx
 Meet our Residency Program Director
Meet our Residency Program Director