X chromosome switch offers hope for girls with Rett syndrome
By Nadine A Yehya
Study successfully tested new gene therapy in X-linked childhood brain conditions
(SACRAMENTO)
Researchers led by UC Davis Health scientist Sanchita Bhatnagar have developed a promising gene therapy that could treatRett syndrome. The therapy works on reactivating healthy but silent genes responsible for this rare disorder and possibly other X-linked conditions, such as fragile X syndrome.
Rett syndrome is a genetic condition that affects mostly girls. It is caused by a defective MECP2 gene located on the X chromosome. This gene contains instructions for the synthesis of MeCP2 protein.
Girls with Rett syndrome may have too little of this protein or their protein may not work properly. This protein deficiency can cause a range of symptoms, including loss of speech, impaired hand movements, breathing difficulties and seizures.
Silenced genes
Sanchita Bhatnagar, an associate professor with the UC Davis Department of Medical Microbiology and Immunology.
Females have two X chromosomes (XX). In each cell, one X chromosome will randomly be silenced in a process known as X chromosome inactivation (XCI). In girls with Rett syndrome, the silenced chromosome carries a healthy copy of MECP2.
“Our study looked at reactivating the silenced X chromosome carrying the healthy gene. It showed that reactivating the gene is possible and can reverse the symptoms,” said Bhatnagar, the study’s senior author.
Sponge-like molecules to overcome microRNA’s silencing power
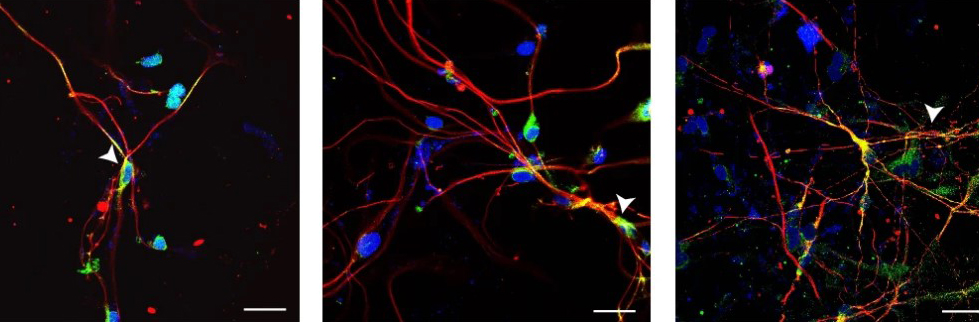
The new study did a genome-wide screening to identify small RNA molecules (microRNA) involved in XCI and X-linked gene silencing. It found that microRNA-106a (miR-106a) was active in switching off X chromosomes and the MECP2 gene.
The team tested if blocking miR-106a could weaken the silencing effect and “wake up” the dormant healthy gene. For that, they used a female mouse model of Rett syndrome and a gene therapy vector developed by ProfessorKathrin Meyerat the Nationwide Children’s Hospital. The vector delivered a special DNA-based molecule that acts as a “sponge” by attracting miR-106a. The molecule reduces the availability of miR-106a at the X chromosome, which provides a therapeutic window for gene activation and MeCP2 production.
Impressive results
The results were very impressive: The treated mice lived longer and showed better movement and cognition than the untreated ones. The study also showed a significant improvement in the breathing irregularities of the treated mice.
“The diseased cell holds its own cure. With our technology, we are just making it aware of its ability to replace the faulty gene with a functional gene,” Bhatnagar explained. “Even a small amount of this gene expression (activation) has therapeutic benefit.”
Importantly, the Rett mouse model handled the treatment well.
“Our gene therapy-based approach targeting X chromosome silencing showed significant improvement of several symptoms of Rett syndrome,” Bhatnagar said. “Girls with Rett exhibit a wide range of symptoms, limited mobility and communication skills. They have apnea and seizures. It would be life-changing if we can help reverse some of their symptoms so they can speak if they're hungry or walk to get a drink. What if we can prevent these seizures and apnea episodes, or simply reduce them?”
Rett syndrome still has no cure. For families affected by Rett syndrome, this discovery brings some hope that a treatment could one day change lives. This approach could also work for similar conditions caused by X-linked genes.
Before moving to clinical trials, the researchers need to conduct safety studies to further evaluate the potency of the treatment and the right dosage.
For a complete list of authors, please checkthe article. This study was funded by Alcyone Therapeutics Inc. and the Hartwell Foundation.