New study links muscle scarring to stem cells’ sensitivity to stiffness
(SACRAMENTO)
A UC Davis Health team is on a mission to solve a key mystery in the formation of muscular fibrosis. The researchers are studying why special stem cells known as fibro-adipogenic progenitors (FAPs) get derailed from normal muscle regeneration following injury, and instead produce excess material that can lead to fibrosis. The team may have unlocked a way to prevent these cells from getting stuck in an endless loop of collagen production, causing fibrotic muscles.
Lucas Smith, assistant professor in the Departments of Physical Medicine and Rehabilitation and Neurobiology, Physiology and Behavior
Fibrosis and the resulting scar tissue happen when an injury doesn’t heal properly. It is a major cause of muscle weakness and a hallmark of aging, severe chronic muscle injuries, and muscular dystrophy diseases. Fibrosis prevents muscle regeneration after injury and increases the risk of re-injury.
Smith and his team in the MyoMatrix Lab study the fibrotic process and its link to muscle diseases, such as muscular dystrophies. In their study published in Scientific Reports, they explain the relationship between connective tissue structure, the muscle’s mechanical function and the stem cells’ regenerative capacity.
The matrix holding the muscle together
The extracellular matrix (ECM) is the connective tissue that provides a structure holding myofibers, blood capillaries and nerves in a muscle. It has a key role in the maintenance and repair of muscle fibers.
A fibrotic matrix has different mechanical and architectural properties compared to a healthy matrix. Excessive connective tissue deposits replace normal tissues, causing permanent scarring. Collagen, specifically type I, makes up a large portion of this matrix. An increase in collagen puts the muscle at a higher risk of developing fibrosis.
Understanding the mechanism of muscular fibrosis
The Smith team has a theory about what might be causing fibrosis.
They expected that a stiff environment due to a high collagen level could turn the progenitors into myofibroblasts that produce collagen. More collagen makes the matrix even more stiff. This leads to an endless positive feedback loop.
“This self-feeding loop had never really been looked at, particularly in skeletal muscles,” Smith said. “We wanted to know if it’s just the stiffness driving that loop, or is it the other matrix components with their alignments and bonding and the fibril size?”
The researchers needed to answer: Does FAPs’ sensitivity to their environment play a role in the development of regenerative versus fibrotic tissue?
To study the sensitivity of FAPs, they looked at the progenitors’ response to changes in their mechanical environment and the matrix architecture.
The team collaborated with Kent Leach, a professor of orthopedic surgery, to fabricate collagen hydrogels to mimic the architectural features of a fibrotic matrix.
They measured the FAPs’ response to mechanical stimulation by examining the location of the Yes-associated protein (YAP), a critical regulator of a cell’s transition to a pro-fibrotic myofibroblast. In its inactive form, YAP resides in the cytoplasm of the stem cells. When activated, it moves to the nucleus to trigger the gene responsible for cell division and death.
They also looked at whether FAPs turned into myofibroblasts – the primary ECM-secreting cells during wound healing and fibrosis. Their activation is key in tissue repair and central to developing posttraumatic stiffness.
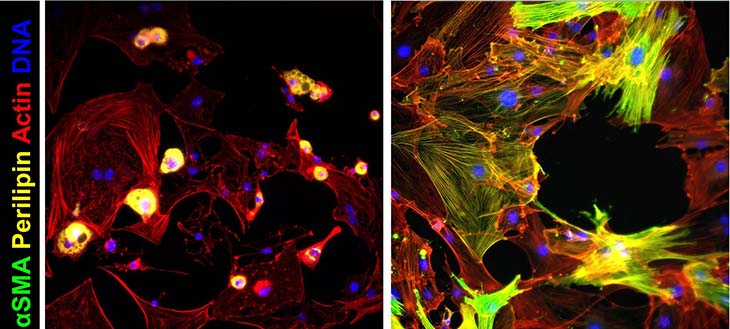
Smooth muscle alpha-actin (αSMA) and perilipin proteins in fat cells (left) and FAP (right) media
Findings
First, the researchers assessed FAPs’ response to changes in matrix stiffnesses. In stiff environments, the Yes-associated protein enters the nucleus and drives the production of the fibrotic response. They theorized that FAPs were detecting this protein, and sensing the stiffness of the matrix, which would help explain the positive feedback and progressive nature of muscle fibrosis.
The team found an increase in the protein in the progenitors’ nuclei in stiff substrates. This pointed to FAPs’ ability to detect their underlying substrate mechanics.
“Our study established that stiffness sensing by FAPS is key in triggering the positive feedback loop of fibrosis in skeletal muscle,” said Taryn Loomis, a doctoral candidate at UC Davis and lead author of the study.
The team also tested the effect of the matrix architecture on myofibroblast activation and YAP localization.
They found that the presence of much more collagen – a characteristic of fibrosis – alone did not trigger the myofibroblast activation. This further implicates stiffness as the key regulator of FAPs.
The researchers also evaluated the effect of collagen crosslinking – the chemical bonding of collagens with nearby fibrils – on the progenitors’ behavior. To their surprise, their experiment showed that the crosslinking blocks the activation of the progenitors into myofibroblasts without impacting the protein’s location.
“A crosslinking inhibitor on its own may not be the most straightforward way to prevent the buildup of these pathologic cells,” Loomis said.
Blocking this stiffness sensing may prevent the loop
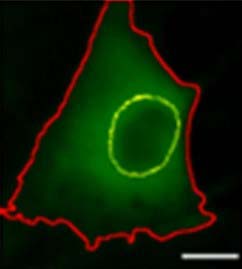
YAP outside nucleus in FAPs cultured in verteporfin for 2 days. Yellow outline indicates nucleus and red outline indicates cytoplasm
The team tested verteporfin, a drug that can block the progenitors’ stiffness sensing. When applied to the cells, the drug prevents YAP from entering the nucleus and activating the myofibroblasts. For the first time, verteporfin was used in the muscle-relevant system and with the progenitor cells. The study, performed on mouse skeletal muscle tissues, showed that this drug might effectively prevent stiffness sensing.
“We're making progress in understanding what's happening at the cellular level and now have a perspective on how we could break that degenerative cycle,” commented Smith. “More testing is needed before we can safely move to clinical trials on verteporfin to prevent fibrosis.”
Coauthors are Lin-Ya Hu, Ross Wohlgemuth, Rosemary Chellakudam and Pooja Muralidharan. This work was supported by a National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) grant (R00AR067867).